Computational Study of 3-Pyridine Carboxaldehyde
1Department of Physics, Univeristy College, Thiruvananthapuram, India.
2Department of Physics, S.N.College, Chempazhanthy, Thiruvananthapuram, India.
3Department of Physics, Govt. Arts and Science College, Calicut, Kerala, India.
4Department of Physics, Fatima Mata National College, Kollam - 691 001, India.
5Department of Physics, TKM College of Arts and Science, Kollam - 691 005, India.
DOI : http://dx.doi.org/10.13005/msri/070145
Download this article as:
![]()
The vibrational wavenumbers of 3-pyridine carboxaldehyde were calculated using Gaussian03 software package at different levels and the fundamental modes are assigned. The predicted infrared and Raman activities are reported. The first hyperpolarizability is calculated and the title compound is an attractive object for future studies of non linear optics. The calculated wavenumbers are in agreement with the reported experimental values.
KEYWORDS:HF; DFT calculations; Hyperpolarizability
Introduction
Pyridine has been extensively studied spectroscopically, due to its applications in many chemical structures of high interest in a variety of biomedical and industrial fields.1 Pyridine has the intrinsic interest of being the azine nearest to benzene. 3-pyridine carboxaldehyde is used in the preparation of 4H-chromines and analogs as activators of caspases and induces apoptosis in human leukemia cells and used as antirheumatic agents, antitumor agents, in the treatment of melanoma, neuroblasoma, insulinoma and colon carcinoma.2 The title compound is widely used in the preparation of pyrido[4,3-g]quinoline–a photochromic compound which is useful for recording and as memory materials, light sensitive materials for radar and display devices.3 Nicotinic acid derivatives are used as antipruritic and for the prevention and treatment of acne vulgaris.4,5 Jose and Mohan6 reported the IR and Raman investigations of the title compound. In the present study, we have calculated the vibrational wavenumbers by using HF and DFT methods and compared with the reported experimental values.
Computational Details
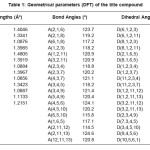
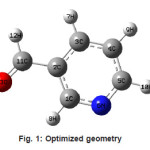
Calculations of the title compound were carried out with Gaussian03 program7 using the Hartree-Fock and DFT (B3LYP) levels of theory using the standard 6-31G* set to predict the molecular structure and vibrational wavenumbers. Molecular geometry (Figure 1) was fully optimized by Berny’s optimization algorithm using redundant internal coordinates. Harmonic vibrational wavenumbers were calculated using the analytic second derivatives to confirm the convergence to minima of the potential surface. The wavenumber values computed contain known systematic errors and hence we have used scaling factors 0.8929 and 0.9613 for HF and DFT methods.8 The absence of imaginary wavenumbers of the calculated vibrational spectrum confirms that the structure deduced corresponds to minimum energy. The optimized geometrical parameters (DFT) are given in Table 1.
Table 1: Geometrical parameters (DFT) of the title compound
 Click on image to enlarge |
Figure 1: Optimized geometry
 Click on image to enlarge |
Results and Discussion
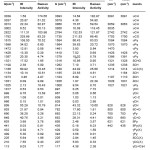
The calculated scaled wavenumbers, experimental wavenumbers given by Jose and Mohan6 and the assignments are given in Table 2. The pyridine CH stretching vibrations are usually observed in the range9 3100-3000 cm-1 and in the present case the DFT calculations give υCH in the range 3056-3093 cm-1. Pyridine ring stretching vibrations10,11 υPy occur in the region 1600-1300 cm-1. These modes involve stretching and contraction of all the bonds in the ring and interaction between the stretching modes. The bands at 1584, 1564, 1461, 1419 and 1379 cm-1 (DFT) are assigned as υPy modes. DFT calculations give the ring breathing mode at 1000 cm-1 as expected.12 The pyridine CH in-plane and out-of-plane deformations are assigned at 1262, 1185, 1104, 1026 cm-1 and at 997, 967, 928, 814 cm-1, respectively.10,11,13
Table 2: Calculated (scaled) wavenumbers and assignments
 Click on image to enlarge |
The aldehyde CH stretching mode14 is assigned at 2794 cm-1 which is expected in the range 2895-2650 cm-1. The aldehyde CH vibrations are assigned at 1316, 939 cm-1 and C=O modes are assigned at 1735 cm-1 (stretching), 782, 652 cm-1 (deformations). The pyridine ring deformations, in-plane, out-of-plane and substituent sensitive vibrations are also identified (Table 2).
Analysis of organic molecules having conjugated π-electron systems and large hyperpolarizability using infrared and Raman spectroscopy has been evolved as a subject of research.15 The potential application of the title compound in the field of non linear optics demands the investigation of its structural and bonding features contributing to the hyperpolarizability enhancement, by analyzing the vibrational modes using the IR and Raman spectrum. The calculated first hyperpolarizability of the title compound is 2.13 10-30 esu, which is comparable with the reported values of similar derivatives.16 We conclude that the title compound is an attractive object for future studies of non linear optics.
References
- Katrizky, A.R., Rees, C.W., and Scriven, E.F.V., (Eds.), Comprehensive Heterocyclic Chemistry II, vol.5,6 Pergamon (1996).
- Xiong, C.S., Hong, Z., Sonychan, I., ans Richard, S., PCT Int. Appl. 22: 139 (2002).
- Shigeru, N, and Takahiro, H, Jpn. Kokai Tokkyo Kobo 11: 5 (1991).
- Mariko, H., Shintaro, I., Yasushi, K., and Tomomi, Y., Jpn. Kokai Tokkyo Koho 22:8 (2002).
- Josef, K., Ger. Offen. 34: 19 (1979).
- Jose, S.P., and Mohan, S., Spectrochim. Acta 64A: 205 (2006).
- Frisch, M.J., et al. Gaussian03, Revision C.02, Gaussian Inc., Wallingford CT (2004).
- Foresman, J.B., Frisch, E., in: Frisch, E;, (Ed.) Exploring Chemistry with Electronic Structure Methods, A Guide to Using Gaussian, Gaussian, Pittsburg, PA (1996).
- Varghese, H.T., Panicker, C.Y., Anto, P.L, and Philip, D., Asian J. Chem. 19: 2627 (2007).
- Roeges, N.P.G., A Guide to the Complete Interpretation of Infrared Spectra of Organic Structures, Wiley, New York (1994).
- Silverstein, R.M, and Webster, F.X., Spectrometric Identification of Organic Compounds, ed. 6, Wiley, Asia (2003).
- Urena, F.P., Gomez, M.F., Gonzalez, J.J.L., and Torres, E.M., Spectrochim. Acta 59A: 2815 (2003).
- Colthup, N.B., Daly, L.H., and Wiberly,S.E.,Introduction to Infrared and Raman Spectroscopy, ed. 2, Academic Press, New York (1985).
- Saier, E.I., Cousins, I.R., and Bassilla, M.R., Anal Chem. 34: 824 (1962).
- Tommasini, M., Castiglioni, C., Del Zoppo, M., and Zerbi, G., J. Mol. Struct. 480: 179 (1999).
- Varghese, H.T., Panicker, C.Y., Madhavan,V.S., Mathew, S., Vinsova, J., and Van Alsenoy, C., J. Raman Spectrosc. doi:10.1002/jrs.2265.
Accepted on: 05 Feb 2010








![]()
![]()
![]()
![]()
