Vibrational Spectroscopic Studies of 2-Amino Picoline
1Department of Physics, Univeristy College, Thiruvananthapuram, India.
2Department of Physics, Govt. Arts and Science College, Calicut, India.
3Department of Physics, S.N.College, Chempazhanthy, Thiruvananthapuram, India.
4Department of Physics, Fatima Mata National College, Kollam - 691 001, India.
5Department of Physics, TKM College of Arts and Science, Kollam - 691 005, India.
DOI : http://dx.doi.org/10.13005/msri/070138
Download this article as:
![]()
The vibrational wavenumbers of 2-amino picoline were calculated using Gaussian03 software package at different levels and the fundamental modes are assigned. The predicted infrared and Raman activities are reported. The first hyperpolarizability is calculated and the 2-amino picoline is an attractive object for future studies of non linear optics. The calculated wavenumbers are in agreement with the reported experimental values.
KEYWORDS:HF; DFT calculations; Hyperpolarizability; Amino
Introduction
Pyridine has been extensively studied spectroscopically, due to its application sin many chemical structures of high interest in a variety of biomedical and industrial fields.1 Pyridine has the intrinsic interest of being the azine nearest to benzene. Amino pyridines attracts the attention of many spectroscopists due to their wide application in pharmacology and agro chemistry. They serve as a good anesthetic agent and hence are used in the preparation of drugs for certain brain disease. The derivatives of picoline has potent hypolipidemic effects, antineoplastic and anti-inflammatory activities and show good activity against lukemia and human glioma cell growth.2 Jose and Mohan3 reported the vibrational spectra and normal co-ordinate analysis of 2-amino picoline. Ab initio quantum mechanical method is at present widely used for simulating the IR spectrum. Such simulations are indispensable tools to perform normal coordinate analysis that modern vibrational spectroscopy is unimaginable without involving them. In the present study, we have calcualted the vibrational wavenumbers of the title compound by using Hartree-Fock and DFT methods and compared with the IR and Raman bands observed by Jose and Mohan.3 Many organic molecules containing conjugated ð electrons and characterized by large values of molecular first hyperpolarizabilities have been analyzed by means of vibrational spectroscopy.4 In this context, the hyperpolarizability of the title compound was calculated theoretically.
Computational Details
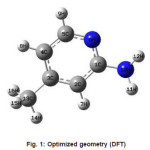
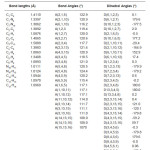
Calculations of the title compound were carried out with Gaussian03 program5 using the Hartree-Fock and DFT (B3LYP) levels of theory using the standard 6-31G* set to predict the molecular structure and vibrational wavenumbers. Molecular geometry (Figure 1) was fully optimized by Berny’s optimization algorithm using redundant internal coordinates. Harmonic vibrational wavenumbers were calculated using the analytic second derivatives to confirm the convergence to minima of the potential surface. The wavenumber values computed contain known systematic errors and hence we have used scaling factors 0.8929 and 0.9613 for HF and DFT methods.6 The absence of imaginary wavenumbers of the calculated vibrational spectrum confirms that the structure deduced corresponds to minimum energy. The optimized geometrical parameters (DFT) are given in Table 1.
Figure 1: Optimized geometry (DFT)
 Click on image to enlarge |
Table 1: Geometrical parameters (DFT) of the title compound
 Click on image to enlarge |
Results and Discussion
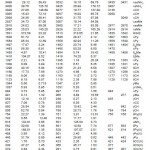
The calculated scaled wavenumbers, experimental wavenumbers given by Jose and Mohan3 and the assignments are given in Table 2. The NH2 stretching modes are expected in the region7 3250-3480 cm-1 and the DFT calculation give 3532 and 3425 cm-1 as asymmetric and symmetric NH2 stretching modes. Jose and Mohan3 reported bands at 3435, 3431 and 3407 cm-1 as NH2 stretching modes. The NH2 scissoring vibrations, expected7 around 1650 cm-1 appear at 1642 cm-1 Raman spectrum and at 1635 cm-1 in the IR spectrum. The DFT calculations give this mode at 1613 cm-1. The δNH2 scissoring vibrations are reported at 1629 cm-1 for sulfanilamide8 and at 1637 cm-1 in IR , 1634 cm-1 in Raman and 1642 cm-1 in HF for orthanilic acid.9 According to Roeges7 ρ/τNH2 vibration is expected in the region 1070 ± 50 cm-1 and in the present case the DFT calculation give this mode at 1031 cm-1. Kurt et al.10 observed the ωNH2 vibration at 667 cm-1 in the IR spectrum and at 695 cm-1 theoretically. Tzeng et al.11 calculated the wavenumber of the wagging vibration of amino group at 649 cm-1 and experimentally at 665 cm-1. For the title compound the DFT calculation give the wagging mode of amino group at 601 cm-1. Primary aromatic amines with nitrogen directly attached to the ring absorb in the region 1330-1260 cm-1 due to the stretching of the ring carbon-nitrogen bond.11.12 The band observed at 1276 cm-1 in IR, 1266 cm-1 in Raman and 1273 cm-1 (DFT) is assigned as C-N stretching mode.
The asymmetric stretching vibrations of the methyl group7,12 are expected in the range 2905-3000 cm-1 and symmetric methyl stretching vibration in the range 2860-2900 cm-1. In the present case the DFT calculation give the methyl stretching modes at 3007, 2980 and 2929 cm-1 which are in agreement with the reported values.3 The methyl deformation bands are expected in the range7,12 1400-1500 cm-1 and the bands at 1482, 1454, 1419 cm-1 given by DFT is assigned as these modes. The methyl rocking vibrations7 are expected around 1045 cm-1 and in the range 970 ±70 cm-1. In the present case ρMe are assigned at 1047 and 965 cm-1 theoretically. The methyl and amino torsions are often assigned in the low wavenumber region.7
The pyridine CH stretching vibrations13-15 are observed in the range 3000-3100 cm-1. Jose and Mohan3 reported CH stretching vibrations at 3135, 3059 cm-1 in the IR spectrum and at 3049 cm-1 in the Raman spectrum. The DFT calculations give these modes at 3077, 3057 and 3043 cm-1. The pyridine ring stretching vibrations16 occur in the general region 1600-1300 cm-1. These vibrations involve stretching and contraction of all the bonds in the ring and interaction between the stretching modes. In the present case the DFT calculations give υPy modes at 1596, 1558, 1465, 1385, 1316 cm-1. The pyridine ring breathing mode is assigned at 997 cm-1 (DFT).17 The in-plane and out-of-plane CH deformations are expected above 1000 and below 1000 cm-1 and all these bands (Table 2) are assigned.7
Table 2: Calculated (scaled) wavenumbers and assignments
 Click on image to enlarge |
Analysis of organic molecules having conjugated π-electron systems and large hyperpolarizability using infrared and Raman spectroscopy has been evolved as a subject of research.18 The potential application of the title compound in the field of non linear optics demands the investigation of its structural and bonding features contributing to the hyperpolarizability enhancement, by analyzing the vibrational modes using the IR and Raman spectrum. The calculated first hyperpolarizability of the title compound is 2.47×10-30 esu, which is comparable with the reported values of similar derivatives.19 We conclude that the title compound is an attractive object for future studies of non linear optics.
References
- Katrizky, A.R., Rees, C.W., and Scriven, E.F.V., (Eds.), Comprehensive Heterocyclic Chemistry II, vol.5,6 Pergamon (1996).
- Das, M.K., Maiti, P.K., Roy S.,, Mittakanli, M., Morse, K.W., and Hall, I.H., Arch. Pharm. Weinheim 325: 267 (1992).
- Jose, S.P., and Mohan, S., Spectrochim. Acta 64A: 240 (2006).
- Anto, P.L.,Anto, R.J., Varghese, H.T., Panicker, C.Y., and Philip, D., J. Raman Spectrosc. doi. 10.1002/jrs.2406.
- Frisch, M.J., et al. Gaussian03, Revision C.02, Gaussian Inc., Wallingford CT (2004).
- Foresman, J.B., Frisch, E., in: Frisch, E;, (Ed.) Exploring Chemistry with Electronic Structure Methods, A Guide to Using Gaussian, Gaussian, Pittsburg, PA (1996).
- Roeges, N.P.G., A Guide to the Complete Interpretation of Infrared Spectra of Organic Structures, Wiley, New York (1994).
- Varghese, H.T., Panicker, C.Y., Anto, P.L., and Philip, D., J. Raman Spectrosc. 37: 487 (2006).
- Anto, P.L., Panicker, C.Y., Varghese, H.T., and Phillip, D., J. Raman Spectrosc. 37: 1265 (2006).
- Kurt, M., Yurdakul, M., and Yurdakul, S., J. Mol. Struct. Theochem. 711: 25 (2004).
- Tzeng, W.B., Narayanan, K., Sheih, K.C., and Tung, C.C., J. Mol. Struct. Theochem. 428: 231 (1998).
- Colthup, N.B., Daly, L.H., and Wiberly,S.E., Introduction to Infrared and Raman Spectroscopy, ed. 2, Academic Press, New York (1985).
- Klots, T.D., Spectrochim. Acta 54A: 1451 (1998).
- Walters, V.A, Snavely, D.L., Colson, S.D., Wilberg, K.B., and Wong, K.N., J. Phys. Chem. 90: 592 (1986) 592.
- JE.Arenas, J.E., Ottero, J.C., Centeno, S.P, Tocon, I.L, and Soto, J., Surf. Sci. 511: 163 (2002).
- Silverstein, R.M, and Webster, F.X., Spectrometric Identification of Organic Compounds, ed. 6, Wiley, Asia (2003).
- Urena, F.P., Gomez, M.F., Gonzalez, J.J.L., and Torres, E.M., Spectrochim. Acta, 59A: 2815 (2003) 2815.
- Tommasini, M., Castiglioni, C., Del Zoppo, M., and Zerbi, G., J. Mol. Struct. 480: 179 (1999).
- Varghese, H.T., Panicker, C.Y., Madhavan, V.S., Mathew, S., Vinsova, J., and Van Alsenoy, C., J. Raman Spectrosc. doi:10.1002/jrs.2265.
Accepted on: 02 Feb 2010








![]()
![]()
![]()
