C. Yohannan Panicker1*, Hema Tresa Varghese2, Sheena Mary Y3, B. Harikumar4, G. Krishnakumar5, K. Raju3 and P. S. Amala Devi6
1Department of Physics, TKM College of Arts and Science, Kollam - 69 1005, India.
2Department of Physics, Fatima Mata National College, Kollam - 691 001, India.
3Department of Physics, Univeristy College, Thiruvananthapuram, India.
4Department of Chemistry, TKM College of Arts and Science, Kollam - 691 005, India.
5Department of Physics, College of Engineering, Thiruvananthapuram, India.
6Department of Physics, S.N.College, Chempazhanthy, Thiruvananthapuram, India.
DOI : http://dx.doi.org/10.13005/msri/070140
Article Publishing History
Article Received on : 15 Nov 2009
Article Accepted on : 20 Dec 2009
Article Published :
Plagiarism Check: No
Article Metrics
ABSTRACT:
The vibrational wavenumbers of guanidinium hydrogenselenate were calculated using Gaussian03 software package at different levels and the fundamental modes are assigned. The predicted infrared and Raman activities are reported. The first hyperpolarizability is calculated and the guanidinium hydrogenselenate is an attractive object for future studies of non linear optics. The calculated wavenumbers and geometrical parameters are in agreement with the reported experimental values.
KEYWORDS:
Guanidinium hydrogenselenate; HF; DFT calculations; Hyperpolarizability
Copy the following to cite this article:
Panicker C. Y, Varghese H. T, Mary Y S. , Harikumar B, Kumar G. K, Raju K, Devi P. S. A. Vibrational Spectroscopic Studies of Guanidinium Hydrogenselenate C(NH2)3.Hseo4. Mat.Sci.Res.India;7(1)
|
Copy the following to cite this URL:
Panicker C. Y, Varghese H. T, Mary Y S. , Harikumar B, Kumar G. K, Raju K, Devi P. S. A. Vibrational Spectroscopic Studies of Guanidinium Hydrogenselenate C(NH2)3.Hseo4. Mat.Sci.Res.India;7(1). Available from: http://www.materialsciencejournal.org/?p=2333
|
Introduction
Guanidinium complexes are biologically important due to their presence as a functional group in amino acids.1-5 Their physical properties are of considerable interest with regard to the variety of applications in the field of ferroelectricity, biotechnology, medicine, etc. Bushiri et al.5,6 and Fleck et al.7 reported the structural and spectral studies of hydrated transition metal guanidinium compounds. Among the transition metal guanidinium complexes, zinc compound is the only anhydrous guanidinium sulphate.7,8 Antony et al.9 reported the spectroscopic properties of guanidinium zinc suphate and ab initio calculations. The guanidinium ion, is relatively simple chemical species whose structure is related to those of amides and proteins in which there is considerable interest. Two part compound with organic cation and inorganic anion are intensively investigated. Recently many new compounds have been obtained for using in nonlinear optics. Usually, these complexes are very stable and have good nonlinear optical properties. Good chemical and physical stability is connected with the existence of inorganic part in investigated compounds, whereas the organic part is responsible for non linear optical properties. Drozd et al.10 reported the crystal structure, differential scanning calorimetry and vibrational low temperature investigations of the title compound. In the present work, we have calculated the geometrical parameters, vibrational wavenumbers and hyperpolarizability theoretically using Gaussian03 set of chemical codes and compared with reported values.
Computational Details
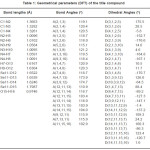
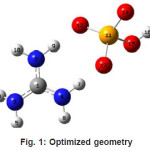
The vibrational wavenumbers were calculated using the Gaussian03 software package on a personal computer.11 The computations were performed at HF/6-31G* and B3LYP/6-31G* levels of theory to get the optimized geometry (Fig. 1) and vibrational wavenumbers of the normal modes of the title compound. DFT calculations were carried out with Becke’s three-parameter hybrid model using the Lee-Yang-Parr correlation functional (B3LYP) method. Molecular geometries were fully optimized by Berny’s optimization algorithm using redundant internal coordinates. Harmonic vibrational wavenumbers were calculated using analytic second derivatives to confirm the convergence to minima in the potential surface. The DFT hybrid B3LYP functional tends also to overestimate the fundamental modes; therefore scaling factors have to be used for obtaining a considerably better agreement with experimental data. Thus, a scaling factor of 0.9613 has been uniformly applied to the B3LYP and 0.8929 for HF methods calculated wavenumbers.12 The observed disagreement between theory and experiment could be a consequence of the anharmonicity and of the general tendency of the quantum chemical methods to overestimate the force constants at the exact equilibrium geometry. The obtained geometrical parameters are given in Tables 1.
Table 1: Geometrical parameters (DFT) of the title compound
Figure 1: Optimized geometry
Results and Discussion
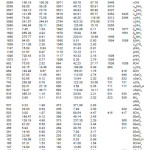
The calculated (scaled) wavenumbers and assignments are given in Table 2. The guanidinium ion vibrations are mainly due to NH and CN bonds. The fundamental modes of CN3 are expected1 at 1041 and 1695, 717 and 498 cm-1. In associated aliphatic and alicylcic primary amines,13 the NH2 asymmetric stretching vibration occurs at 3365±25 cm-1 and symmetric stretching vibtration in the region 3290± 30 cm-1. The NH2 scissoring vibration14 give rise to a broad band in the region 1600 ± 50 cm-1. According to Roeges14 the rocking/twisting NH2 mode is expected in the region 1160 ± 140 cm–1. Drozd et al.10 reported the NH2 vibrations at 3449, 3377, 1564, 1555 cm-1 in the IR spectrum, 3443, 3314, 1558, 1549, 1098 cm-1 in the Raman spectrum. The vibrations due to CN are reported at 1646, 1008, 731 cm-1 in the IR spectrum and at 1661, 731 cm-1 in the Raman spectrum.10 The stretching vibrations of the Se-O bonds15 appear in the region between 1000-800 cm-1. The SeO4 vibrations are reported at 953, 909, 876, 837 cm-1 in the IR spectrum and at 916, 848 cm-1 in the Raman spectrum.10
For the title compound, the DFT calculations give the bond lengths Se11-O14, Se11-O12, Se11-O13, Se11-O15, C1-N2, C1-N3, C1-N4 as 1.6246, 1.6522, 1.6539, 1.7987, 1.3663, 1.3282, 1.3261Å where as the reported values are 1.6055, 1.6095, 1.6204, 1.7124, 1.2878, 1.3079, 1.3358 Å respectively10. Drozd et al.10 reported the bond angles O14-Se11-O12, O14-Se11-O13, O12-Se11-O13, O14-Se11-O15, O12-Se11-O15, O13-Se11-O15, N2-C1-N3, N2-C1-N4, N3-C1-N4 as 116.1, 112.1, 110.5, 103.7, 107.1, 116.5, 120.1, 120.7, 119.5º whereas in the present case the corresponding values are 116.7, 116.0, 110.9, 104.0, 103.4, 116.0, 119.1, 120.4, 120.5º, respectively.
Analysis of organic molecules having conjugated π-electron systems and large hyperpolarizability using infrared and Raman spectroscopy has evolved as a subject of research.16 The first hyperpolarizability (β0) of this novel molecular system is calculated using theoretically, based on the finite field approach. In the presence of an applied electric field, the energy of a system is a function of the electric field. First hyperpolarizability is a third rank tensor that can be described by a 3×3×3 matrix. The 27 components×of the 3D matrix can be reduced to 10 components due to the Kleinman symmetry.17 The calculated first hyperpolarizability of the title compound is 2.8210–30 esu (B3LYP/6-31G* methods). We conclude that the title compound is an attractive object for future studies of non linear optical properties.
Table 2: Calculated (scaled) wavenumbers and assignments
References
- Chakarborty, D., and Manogaran, S., Ind. J. Chem. 33A: 969 (1994).
- Sension, R.J., Hudson, B., and Callis, P.R.,J. Phys. Chem. 94: 4015 (1990).
- Nemec, I., Machackova, Z., Teubner, K.,Cisarova, I., Vanek, P., and Micka, Z., J. Solid State Chem. 177: 4655 (2004).
- Carver, G., Spichiger, D., and Tregenna-Piggot, P.L.W., J. Chem. Phys. 122: 124511 (2005).
- Bushiri, M.J., Antony, C.J., and Fleck, M., J.Raman Spectrosc. 39: 368 (2008).
- Bushiri, M.J., Antony, C.J., and Fleck, Solid State Commun. 143: 348 (2007).
- Fleck, M., Bohar, I., and Tillmanns, E., Solid State Sci., 6: 469 (2004).
- Morimoto, C.N., and Lingafelter, E.C., Acta Cryst. 26B: 335 (1970).
- Antony, C.J., Bushiri, M.J., Varghese, H.T., Panicker, C.Y., and Fleck, M., Spectrochim. Acta doi:10.1016/j.saa/2009.04.024.
- Drozd, M., Baran, J., and Pietraszko, A., Spectrochim. Acta 61A: 2775 (2005)
- Frisch, M.J., et al., Gaussian03, Revision C.02., Gaussian Inc., Wallingford, CT (2004).
- Foresman, J.B., in: Frisch, E., (Ed.),Exploring Chemistry with Electronic Structure Methods: A Guide to Using Gaussian, Pittsburg, PA, (1996).
- Segal, L., and Eggerton, F.V., Appl. Spectrosc. 15: 112 (1961).
- Roeges, N.P.G., A Guide to the Complete Interpretation of Infrared Spectra of Organic Structures, Wiley, New York (1994).
- Paetzold, R., and Amoulong, H., Z. Anorg. Allgem. Chem. 317: 166 (1962).
- Tommasini, M., Castiglioni, C., Del Zoppo, M., and Zerbi, G., J. Mol. Struct. 480: 179(1999).
- Kleinman, D.A., Phys. Rev. 126: 1977 (1962).

This work is licensed under a Creative Commons Attribution 4.0 International License.